La maladie de Huntington est une maladie génétique neurodégénérative. Elle entraîne des troubles moteurs, cognitifs et psychiatriques. Découvrez les différents symptômes, les causes et les traitements possibles de cette pathologie handicapante.
Qu’est-ce que la maladie de Huntington ?
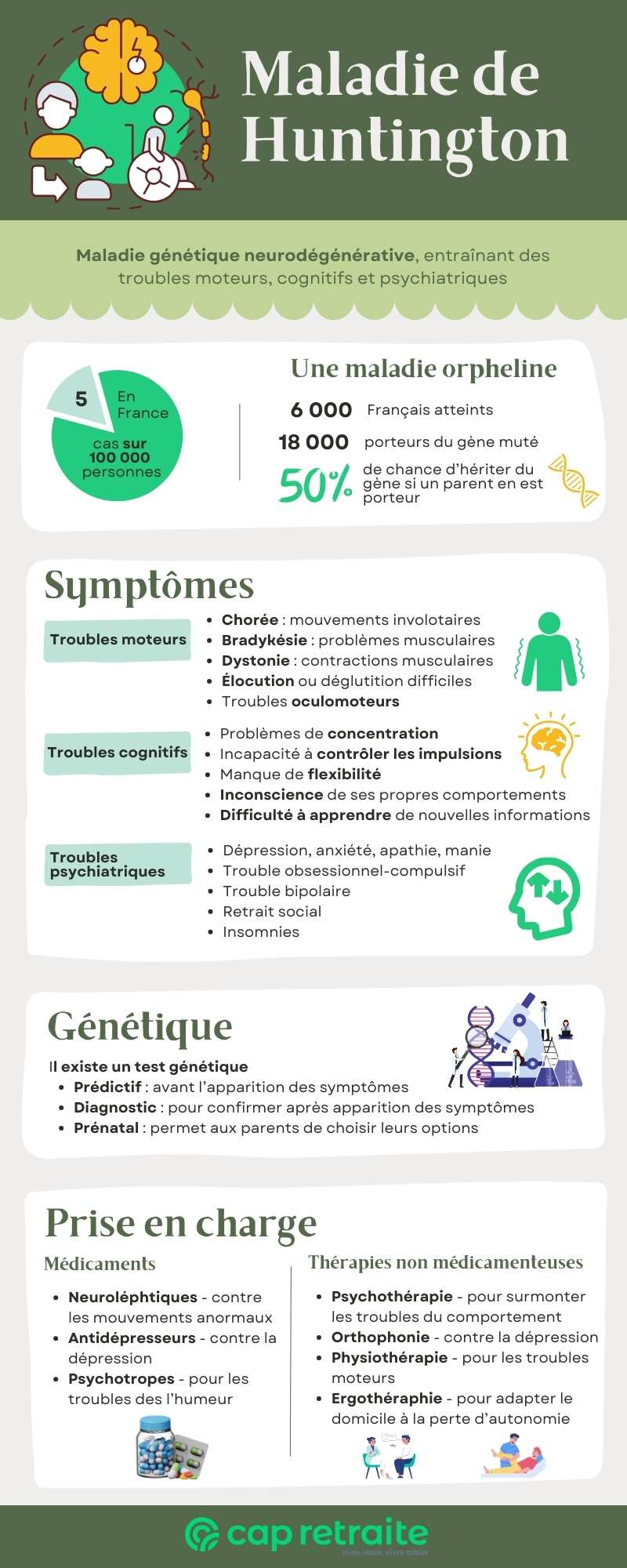
La maladie de Huntington est une maladie neurodégénérative héréditaire. La dégénérescence progressive des cellules du cerveau (neurones) affecte les capacités fonctionnelles du patient. Résultat : des troubles moteurs, cognitifs et psychiatriques se développent au fil de l’évolution de la pathologie.
Il s’agit d’une pathologie rare, dite orpheline. En effet, sa prévalence au sein de la population française est d’environ 5 cas sur 100 000 personnes.
La maladie de Huntington en chiffres (source INSERM) :
- 6 000 Français en sont atteints (présentent des symptômes),
- 18 000 individus sont porteurs du gène muté.
La progression de la pathologie peut s’étaler sur 10 à 25 ans, période pendant laquelle elle tue progressivement les cellules nerveuses du cerveau.
Cette maladie génétique est également appelée chorée de Huntington, en raison de l’un des symptômes les plus visibles, la chorée (voir les symptômes ci-après).
La pathologie se déclare entre 30 et 50 ans, en général. Mais elle peut se produire, plus rarement, à n’importe quel âge de 1 à 80 ans.
Quels sont les symptômes de la maladie de Huntington ?
La chorée de Huntington entraîne en général des troubles moteurs, cognitifs et psychiatriques. Leurs manifestations, étape d’apparition et intensité varient d’un patient à l’autre.
| Type de troubles | Liste des symptômes |
|---|---|
| Troubles moteurs | Mouvements involontaires et déficience des mouvements volontaires : · mouvements saccadés, brusques et involontaires (chorée) · problèmes musculaires, tels que lenteur et rigidité (bradykinésie) · contractions musculaires involontaires (dystonie), · difficulté à initier un mouvement · troubles oculomoteurs (fermeture involontaire des yeux), · démarche, posture et équilibre anormaux · élocution, mastication ou déglutition difficiles |
Troubles cognitifs | · atteinte des fonctions exécutives nécessaires à l’organisation, la hiérarchisation ou la concentration · manque de flexibilité ou tendance à persister dans un même acte, une même pensée… · absence de contrôle des impulsions pouvant entraîner des accès de colère ou des actions irréfléchies · inconscience de ses propres comportements et capacités · lenteur de la mémoire de travail · difficulté à apprendre de nouvelles informations |
| Troubles psychiatriques | · dépression · irritabilité · tristesse ou apathie · retrait social · insomnie · fatigue et perte d’énergie · pensées morbides · trouble obsessionnel-compulsif – pensées intrusives et comportements répétitifs · manie : humeur « élevée » (euphorique), suractivité, comportement impulsif, diminution de la pudeur et estime de soi exagérée · trouble bipolaire – alternance d’épisodes de dépression et de manie |
| Autres | Perte de poids, surtout à un stade avancé |
Quelles sont les causes de la maladie de Huntington ?
La maladie de Huntington est causée par la mutation du gène de la protéine huntingtine, se trouvant sur le chromosome 4 en position 4p16.3.
Cette pathologie se transmet de génération en génération, dans une famille, selon un schéma autosomique dominant, ce qui signifie qu’une copie du gène muté dans chaque cellule suffit pour provoquer la maladie.
Les enfants d’un individu porteur ont une chance sur deux d’avoir la maladie, si l’autre parent n’est pas atteint.
Dans de rares cas, une personne atteinte de la chorée de Huntington n’a pas de parent atteint du trouble.
Comment la maladie de Huntington est-elle diagnostiquée ?
Le diagnostic de la chorée de Huntington repose sur la description des symptômes et l’élimination d’autres maladies neurodégénératives apparentées (notamment les formes génétiques de la maladie d’Alzheimer ou la démence fronto-temporale). Le médecin cherchera aussi la présence d’antécédents familiaux.
Le gène responsable de cette maladie dégénérative a été découvert en 1993. Un test génétique permet donc de confirmer ou d’infirmer le diagnostic chez les personnes dont un parent est porteur de la mutation génétique ou atteint de la chorée de Huntington.
Le test génétique peut également être effectué en l’absence de symptômes, si l’un des parents est atteint. Il peut faire partie du diagnostic prénatal et justifier une interruption de grossesse, si le fœtus est porteur. Un conseil génétique sera alors fournir pour assurer le meilleur accompagnement. Certains couples choisiront d’avoir recours à la fécondation in vitro, pour faire un test génétique et choisir de réimplanter le ou les embryons non porteurs du gène.
Comment soigner la maladie de Huntington ?
À l’heure actuelle, il est impossible de guérir la maladie de Huntington et d’interrompre sa progression.
Il existe néanmoins des traitements symptomatiques. Le but : atténuer les troubles moteurs et psychiatriques. Différentes thérapies permettent d’aider le patient à vivre avec la maladie et à rester autonome aussi longtemps que possible.
Les traitements médicamenteux
Ils doivent être adaptés par un psychiatre ou un neurologue. En effet, le médecin évaluera les bienfaits face aux effets indésirables.
- Les neuroleptiques réduisent les mouvements anormaux : la Tétrabénazine supprime la chorée, mais risque d’aggraver ou entraîner la dépression. Les antipsychotiques réduisent également les mouvements, mais peuvent empirer la dystonie et la rigidité musculaire. Ils sont aussi utilisés pour supprimer les accès de colère et l’agitation. La Rispéridone a en général moins d’effets secondaires.
- Les antidépresseurs peuvent être utilisés pour traiter la dépression.
- Des psychotropes seront souvent prescrits pour stabiliser les problèmes d’humeur, notamment pour le trouble bipolaire.
Les thérapies non médicamenteuses
- La psychothérapie : elle vise à aider la personne à surmonter ses troubles comportementaux et à apprendre des stratégies pour vivre avec la maladie.
- L’orthophonie : l’orthophoniste aide le patient à améliorer son élocution, mais aussi à faire face aux problèmes musculaires entraînant des difficultés à avaler et déglutir.
- La physiothérapie : un kinésithérapeute pourra apprendre au patient à faire des exercices adaptés à ses troubles moteurs, pour renforcer ses muscles et maintenir ses capacités fonctionnelles aussi longtemps que possible. Le kinésithérapeute apprendra également au patient à utiliser un déambulateur, puis un fauteuil roulant.
- L’ergothérapie : l’ergothérapeute a pour rôle d’aider le malade atteint de la maladie de Huntington à adapter son domicile à la perte d’autonomie. Il lui apprendra aussi à utiliser les équipements de mobilité et d’assistance nécessaires aux actes de la vie quotidienne (alimentation, habillage, etc.)
La prise en charge de la chorée de Huntington nécessite la mise en place de stratégies pour aider le patient à vivre avec la perte d’autonomie, qui affecte jusqu’à ses possibilités de s’alimenter. Les aidants seront souvent sollicités ou devront recevoir l’assistance de professionnels de santé ayant bénéficié d’une formation adaptée.
Les patients et familles peuvent trouver de l’aide, notamment, auprès de l’association Huntington France.
Où en est la recherche sur la maladie de Huntington ?
La recherche sur la prise en charge de la maladie de Huntington fait aujourd’hui l’objet d’environ cinquante essais cliniques menés dans le monde entier. Les chercheurs analysent des traitements symptomatiques pour les différents troubles : chorée, ainsi que les symptômes cognitifs et moteurs.
Les approches les plus innovantes visent à freiner l’évolution de la pathologie grâce à la neuroprotection, la thérapie génique et la greffe de neurones.
- La neuroprotection expérimente différentes molécules pour réduire la mort des neurones. Toutefois, aucun médicament efficace n’a encore été identifié.
- La thérapie génique cherche à réguler la manière dont le gène muté est activé dans les cellules à l’aide de plusieurs techniques. Bien que prometteuses, ces méthodes font face à des défis de tolérance et d’efficacité clinique.
- La thérapie cellulaire tente de remplacer les neurones détruits par des greffes. Elle rencontre toutefois des obstacles, tels que des réactions immunitaires et des difficultés d’approvisionnement en cellules fœtales. Les recherches se poursuivent pour surmonter ces limitations.
Questions fréquentes
Peut-on mourir de la chorée de Huntington ?
La chorée de Huntington est une maladie neurodégénérative. En tant que telle, elle entraîne une perte d’autonomie croissante, mais ne cause pas la mort directement.
Les causes de décès des personnes atteintes de la maladie de Huntington sont ses complications. Il s’agit notamment d’infections, comme la pneumonie d’aspiration, ou de maladies cardio-vasculaires, comme la maladie coronarienne et l’infarctus du myocarde.
De plus, les personnes atteintes de cette pathologie ont plus de risques de mourir d’étouffement, de troubles respiratoires, de maladies gastro-intestinales (telles que le cancer du pancréas) ou des suites d’un suicide que la population générale. La perte d’autonomie peut également accroître le risque de chute.
Comment la chorée de Huntington évolue-t-elle ?
On peut définir trois stades de la maladie de Huntington, en fonction des difficultés rencontrées par le patient.
Le stade précoce
Les symptômes sont plus faciles à gérer au début de la maladie. Vous pouvez vous sentir de mauvaise humeur ou maladroit, et avoir des difficultés avec la réflexion complexe. Vous commencerez également à avoir de petits mouvements incontrôlables.
Généralement, à ce stade vous pouvez poursuivre vos activités quotidiennes.
Le stade intermédiaire
Les changements qui s’opèrent sur les plans physique et mental vont rendre impossibles le travail, la conduite et les tâches ménagères. Des difficultés à avaler apparaissent et vous risquez de perdre du poids. Vous avez un moins bon équilibre et le risque de chute augmente.
À ce stade, vous êtes encore capable de prendre soin de vous. En général, vous pouvez faire votre toilette, vous habiller et manger seul ou avec de l’aide.
Le stade final
À un stade avancé, vous allez avoir besoin d’aide pour toutes les activités de la vie quotidienne. Vous serez alité et aurez besoin d’une prise en charge 24 h/24, à domicile ou dans un établissement médico-social.
Quelle espérance de vie avec la maladie d’Huntington ?
La vitesse de progression de la chorée d’Huntington entre ces différents stades varie d’une personne à l’autre. L’espérance de vie après diagnostic est de 10 à 30 ans, selon qu’il s’agit de la forme juvénile (à la progression plus rapide) ou tardive (au développement plus lent).
Comment vivre avec la maladie de Huntington ?
Vous pouvez agir de plusieurs façons pour améliorer votre qualité de vie au fil de l’évolution de la chorée de Huntington.
- Faites une activité physique régulière : la recherche montre que l’exercice physique aide à réduire les symptômes.
- Adoptez une alimentation saine : vous risquez d’avoir besoin de manger plus qu’avant. Vous pouvez en effet brûler jusqu’à 5 000 calories par jour à cause des mouvements involontaires.
- Buvez beaucoup d’eau : lorsque la déglutition devient difficile, vous risquez de facilement vous déshydrater.
- Trouvez un groupe de soutien : demandez à votre neurologue de vous aider à trouver des ressources communautaires. Vous pouvez aussi contacter l’association Huntington France.
- Anticipez votre prise en charge : à un stade plus avancé de la maladie, vous allez avoir besoin de beaucoup d’aide. Renseignez-vous en amont sur les possibilités de mettre en place une aide à domicile. Recherchez en avance une place dans une maison de retraite adaptée. Le personnel soignant est en général formé aux prises en charge spécifiques en Ehpad.
- Nommez votre personne de confiance : au fur et à mesure que la maladie progresse, vous devrez confier à quelqu’un d’autre la prise de décisions importantes.
Certes, vous ne pouvez pas prévenir la maladie de Huntington et il n’existe aucun traitement curatif. Néanmoins, vous pouvez planifier votre prise en charge, car son évolution s’étale sur plusieurs années.
Cet article vous a-t-il été utile ?
Notez cet article afin de nous permettre d’améliorer nos contenus.

Réagissez, posez une question…
Est-ce que un médecin généraliste, peut prescrire des médicaments à qqn qui à les symptômes de la maladie de Hungtinton sans se douter que, c’est de cela que la personne souffre??
Peut-on vraiment donner des médicaments à une personne, sans l’envoyer chez un neurologue pour avoir le bon diagnostic?
Combien de temps cela prend-il pour avoir le résultat des tests?
Bonjour
Je vous remercie pour votre commentaire.
Un médecin généraliste peut prescrire des traitements symptomatiques selon son évaluation clinique, mais la confirmation diagnostique et les délais des tests génétiques dépendent du parcours de soin et de la complexité de chaque situation médicale.
Bonne journée.
Amandine
Ma belle soeur souffre de la maladie de gougereau et de Huntington depuis quelques années elle n’ a pas de chorée mais reste dans sa chambre dans un EHPAD comment cela va t il évoluer pour le moment pas de symptômes particuliers elle a 72 ans
Bonjour
Je vous remercie pour votre commentaire.
L’évolution dépend de nombreux facteurs individuels et médicaux, et un suivi régulier permet d’adapter l’accompagnement si nécessaire.
Bonne journée.
Amandine